原理
総ての物質は原子核と電子から構成されている。様々な物質の特性は系の電子状態に依って決まり、それらの状態はSchrödinger方程式(波動方程式)を解くことに依って得られる。この方程式において、電子の運動エネルギーや電子間Coulomb相互作用などの情報を含むハミルトニアン(𝐻)に対して、基底状態のエネルギー(𝐸)と波動関数(Ψ)を、実験などより得られたパラメータを用いずに解く計算を第一原理計算と言う。ただし、Schrödinger方程式を厳密に解くことは非常に困難なため、種々の近似が用いられる。中でも、基底状態の全エネルギーを電子密度の汎関数で記述する密度汎関数理論(DFT : Density Functional Theory)は、多電子の状態を一電子方程式に簡略化しており、計算の高速化に加え、一定の精度が得られるため、主流な理論となっている。
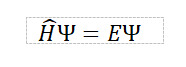
第一原理計算より得られる情報
- 系のエネルギー
- 分子、固体の安定構造、遷移状態構造
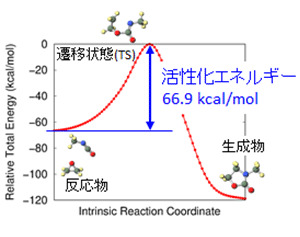
- 化学反応経路、活性化エネルギー
- 各種分光スペクトル(IR, RAMAN, NMR, ESR, XAFSなど)
- 励起エネルギー(吸収スペクトル、発光波長など)
- 溶媒効果
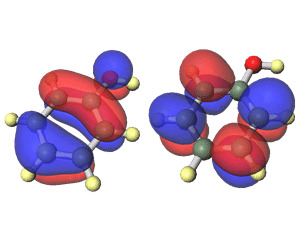
- 波動関数の可視化(結合性、反結合性軌道、HOMO,LUMOの広がりなど)
- 分子、固体中の原子の電荷、スピン分布
- バンドギャップ
- 格子定数、体積弾性率